
Синдром веста клинические рекомендации
Добавил пользователь Алексей Ф. Обновлено: 05.10.2024
Эпилепсия у детей и взрослых

1.1. Идиопатические формы (начало приступов связано с возрастом):
– доброкачественная эпилепсия детского возраста с центрально - темпоральными спайками
– эпилепсия детей с затылочными пароксизмами на ЭЭГ
– первичная эпилепсия чтения.
1.2. Симптоматические формы:
– хроническая прогредиентная парциальная эпилепсия (синдром Кожевникова);
– синдромы со специфическими причинами провокации приступов (рефлекторная эпилепсия);
– лобно-, височно-, теменно-, затылочно-долевая эпилепсия.
1.3. Криптогенные формы (неопределенные формы)
2. ЭПИЛЕПСИЯ И СИНДРОМЫ С ГЕНЕРАЛИЗОВАННЫМИ ПРИСТУПАМИ
2.1. Идиопатические (начало приступов связано с возрастом):
– доброкачественные семейные неонатальные судороги;
– доброкачественные идиопатические неонатальные судороги;
– доброкачественные младенческая миоклоническая эпилепсия;
– эпилепсия с пикнолептическими абсансами (пикнолептическая, абсанс-эпилепсия у детей);
– детская абсансная эпилепсия;
– ювенильная миоклоническая эпилепсия;
– эпилепсия с генерализованными тонико-клоническими судорогами при пробуждении;
– другие формы генерализованной идиопатической эпилепсии;
– эпилепсия со специфическими провоцирующими факторами (рефлекторная и старт-эпилепсия).
2.2 Криптогенные или симптоматические формы (связанные с возрастом появления приступов):
– синдром Веста (инфантильные спазмы);
– синдром Леннокса-Гасто;
– эпилепсия с миоклонически-астатическими приступами;
– эпилепсия с миоклоническими абсансами.
2.3 Симптоматические формы:
2.3.1. Неспецифической этиологии
– ранняя миоклоническая энцефалопатия
– младенческая энцефалопатия с участками изоэлектрической ЭЭГ
– другие симтоматические генерализованные формы эпилепсии
2.3.2 Специфические синдромы
3. ЭПИЛЕПСИЯ И СИНДРОМЫ, НЕОПРЕДЕЛЕННЫЕ ОТНОСИТЕЛЬНО ТОГО, ЯВЛЯЮТСЯ ЛИ ОНИ ФОКАЛЬНЫМИ ИЛИ ГЕНЕРАЛИЗОВАННЫМИ
3.1. Вместе генерализованные и фокальные приступы:
– приступы новорожденных;
– тяжелая миоклоническая эпилепсия раннего детского возраста;
– эпилепсия с длительными пик-волнами на ЭЭГ во время медленной фазы сна;
– синдром афазии-эпилепсии (Ландау-Клеффнера);
– другие неопределенные формы эпилепсии.
3.2. Без определенных генерализованных и фокальных признаков (многие случаи генерализованных тонико-клонических судорог, которые по данным клиники и ЭЭГ нельзя отнести к другим формам эпилепсии данной классификации, а также многие случаи больших судорожных приступов во время сна).
4. СПЕЦИАЛЬНЫЕ СИНДРОМЫ
4.1. Ситуативные (случайные) приступы:
– Фебрильные судороги;
– Приступы, связанные исключительно с острым воздействием метаболических или токсических факторов, а также депривация (лишение) сна, алкоголь, лекарства, эклампсия и т.д.
4.2. Изолированные судороги или изолированный эпилептический статус
Рисунок – 2. Электроклинические синдромы и другие эпилепсии, с группированные по специфичности диагнозов [6].

Диагностика (амбулатория)
ДИАГНОСТИКА НА АМБУЛАТОРНОМ УРОВНЕ
Диагностические критерии
Жалобы: Собираются как у пациента, так и у свидетеля приступа и родственников больного.
Жалобы на пароксизмальные состояния (приступы):
· характер приступа: с утратой сознания, без утраты сознания, судорожные, бессудорожные), частота, продолжительность, наличие ауры;
Анамнез:
· первый или повторный;
· наличие в анамнезе неонатальных и фебрильных приступов;
· наследственной отягощенности по эпилепсии;
· возраст дебюта;
· наличие токсических, гипоксически-ишемических, травматических и инфекционных поражений мозга, включая внутриутробный период;
· нарушения режима приема противосудорожных препаратов;
· нарушение режима труда и отдыха);
· постприступное состояние (описание, видеозапись, ведение дневника приступов).
Физикальное обследование:
· общесоматический статус: общее состояние и его тяжесть, дыхание, пульс, АД, температура тела, измерение массы и роста пациента, окружности головы, осмотр кожных покровов,
· неврологический статус: уровень сознания, общемозговая симптоматика, менингеальные знаки, черепные нервы, двигательно-рефлекторная сфера, чувствительная сфера, координаторная функция, функции тазовых органов, когнитивные функции, вегетативная нервная система, психоэмоциональный статус.
Лабораторные исследования:
· общий анализ крови – при длительном приеме антиэпилептических препаратов, могут возникнуть побочные действия со стороны крови: снижение количества тромбоцитов, снижение свертываемости крови;
· биохимический анализ крови (АЛТ, АСТ, тимоловая проба, билирубин общий амилаза, щелочная фосфатаза, общий белок, мочевина, креатинин (по показаниям) –гепатотоксичность антиэпилептических препаратов необходим контроль биохимических параметров для оценки изменений со стороны печени, поджелудочной железы 2 раза в год;
· исследование мочи общеклиническое (общий анализ мочи) 2 раза в год;
· определение уровня АЭП в крови необходимо:
– низкая эффективность терапии;
– начало антиэпилептической терапии, изменение дозы, изменение торговой версии препарата, изменение сопутствующей терапии;
– оценки побочных эффектов АЭП;
– доказательства стойкого снижения комплаентости терапии;
– беременность, другие сопутствующие заболевания – для коррекции дозы данного препарата в крови.
· определение уровня альфафетопротеина (АФП) – необходимо для раннего выявления врожденной аномалии плода у беременных женщин на 14 – 15 неделе, принимающих АЭП.
Инструментальные исследования:
· Электроэнцефалография – рутинная, позволяет определить наличие патологической электрической активности, указывает на расположение эпилептического очага;
· Длительное ЭЭГ - мониторирование – для уточнения типа приступа и регистрации иктального ЭЭГ;
· МРТ головного мозга – для выявления органической патологии;
· КТ головного мозга – при отсутствии МРТ и при противопоказаниях;
· МРТ головного мозга (по показаниям) ПО эпилептологическому протоколу – метод диагностики входящий в структуру предоперационного обследования, фармокорезистентных формах эпилепсии, для диагностики мезиального темпорального склероза, сосудистых мальформаций, дисгенезий мозга, дифференцировка патологического участка между ФКД и глиальными образованиями, при метаболических, митохондриальных энцефалопатиях и при очагах неясной этиологии;
· Нейросонография;
· УЗИ органов брюшной полости.
Диагностический алгоритм: [7]
Рисунок – 3. Алгоритм действий при пароксизмальных состояниях.

Диагностика (скорая помощь)
ДИАГНОСТИКА НА ЭТАПЕ СКОРОЙ НЕОТЛОЖНОЙ ПОМОЩИ
Диагностические мероприятия: уровень сознания, характер и продолжительность приступа, контроль АД, частоты дыхания, пульс, температуру.
Диагностика (стационар)
ДИАГНОСТИКА НА СТАЦИОНАРНОМ УРОВНЕ
Диагностические критерий на стационарном уровне:
Жалобы и анамнез: см. амбулаторный уровень.
Физикальное обследование: см. амбулаторный уровень.
Лабораторные исследования:
· общий анализ крови – при длительном приеме антиэпилептических препаратов, могут возникнуть побочные действия со стороны крови: снижение количества тромбоцитов, снижение свертываемости крови;
· биохимический анализ крови (глюкозы, АЛТ, АСТ, тимоловая проба, билирубин общий, амилаза, щелочная фосфатаза, общий белок, мочевина, креатинин (по показаниям) – гепатотоксичность антиэпилептических препаратов необходим контроль биохимических параметров для оценки изменений со стороны печени, поджелудочной железы 2 раза в год;
Инструментальные исследования:
· Электроэнцефалография – рутинная, позволяет определить наличие патологической электрической активности, указывает на расположение эпилептического очага;
· Длительное ЭЭГ - мониторирование – для уточнения типа приступа и регистрации иктального ЭЭГ;
· МРТ головного мозга – для выявления органической патологии;
· КТ головного мозга – при отсутствии МРТ;
Диагностический алгоритм: см. амбулаторный уровень.
Перечень основных диагностических мероприятий:
· общий анализ крови с подсчетом тромбоцитов;
· биохимический анализ крови (глюкозы, АЛТ, АСТ, тимоловая проба, билирубин общий, амилаза, щелочная фосфатаза, общий белок, мочевина, креатинин);
· ЭЭГ;
· КТ/МРТ головного мозга;
Перечень дополнительных диагностических мероприятий:
· Нейросонография;
· Электроэнцефалография;
· Видеомониторинг электроэнцефалограммы (1-3-5 часов);
· Ночной видеомониторинг электроэнцефалограммы;
· УЗИ органов брюшной полости;
· МРТ головного мозга (по показаниям) ПО эпилептологическому протоколу – метод диагностики входящий в структуру предоперационного обследования, фармокорезистентных формах эпилепсии, для диагностики мезиального темпорального склероза, сосудистых мальформаций, дисгенезий мозга, дифференцировка патологического участка между ФКД и глиальными образованиями, при метаболических, митохондриальных энцефалопатиях и при очагах неясной этиологии
· Исследование спинномозговой жидкости (по показаниям): при подозрении на инфекцию или опухоль мозга;
· Определение лактатдегидрогиназы (ЛДГ);
· Определение кальция (Ca) в сыворотке крови;
· Определение лактата (молочной кислоты) в сыворотке крови;
· определение уровня АЭП в крови;
· определение уровня альфафетопротеина (АФП).
· общий анализ мочи 2 раза в год;
· определение уровня АЭП в крови;
Синдром Веста – лечение в Германии
Патологии головного мозга и центральной нервной системы значительно снижают качество жизни человека, приводят к возникновению тяжелых нарушений. Среди заболеваний данной группы особенно серьезными являются нарушения врожденного типа или развивающиеся в первые годы жизни. Они сопровождаются отставаниями в развитии и влияют на здоровье человека в будущем. К числу таких патологических процессов относится Синдром Веста.
Диагностика и лечение этого заболевания в клиниках Вивантес проводится только у детей.
На базе клиник концерна Вивантес диагностика и лечение Синдрома Веста в Германии осуществляются врачами высшей категории. Специалисты нашей клиники выполняют раннюю диагностику заболевания, используя в своей практике современное оборудование экспертного класса.
Лечение болезни проводится согласно инновационным методикам, оно включает преимущественно консервативную терапию, но при наличии соответствующих показаний выполняются оперативные вмешательства различной степени сложности.
Что такое Синдром Веста
В неврологии Синдромом Веста называется патологический процесс не воспалительной природы, затрагивающий структуры головного мозга. Данное заболевание характеризуется как младенческая эпилептическая энцефалопатия. Его основной отличительной чертой являются спастические мышечные сокращения, схожие с эпилептическими приступами.
Особенно часто развивается Синдром Веста у детей в первые годы жизни. Согласно статистике, манифестация заболевания происходит в первый год жизни ребенка примерно в 90% случаев. В оставшихся 10% случаев первые признаки болезни проявляются до 4-летнего возраста. Но в то же время у подавляющего большинства детей, спастические спазмы проходят к 3 годам жизни, после чего происходит их трансформация в различные формы эпилепсии.
Синдром Веста относится к группе редких неврологических нарушений. По данным статистики заболевание составляет порядка 2% от всех случаев эпилепсии. Однако если говорить о младенческих формах эпилепсии и эпилептических энцефалопатий, среди них Синдром Веста составляет порядка четверти всех случаев.
Причины развития Синдрома Веста у детей довольно многочисленны, из их числа наиболее распространенными в неврологии считаются:
- последствия перенесенных внутриутробных инфекций;
- гипоксия плода на различных стадиях внутриутробного развития;
- преждевременные роды;
- полученные в процессе родовой деятельности внутричерепные травмы;
- нарушения дыхательной деятельности у младенца;
- некоторые формы аномалий строения головного мозга.
Также важно понимать, что примерно в 13% случаев истинная причина возникновения Синдрома Веста у грудничка остаются неустановленными.
Симптомы и диагностика Синдрома Веста
Как было сказано ранее, в большинстве случаев патологический процесс дебютирует на первом году жизни ребенка. При этом клиническая картина становится более разнообразной по мере развития Синдрома Веста, а начинается прогрессирование патологии преимущественно с задержки психомоторного развития. К числу наиболее характерных клинических признаков специалисты относят:
- выпадение хватательного рефлекса у младенца;
- расстройства фиксации, фокусировки взгляда;
- ребенок не может следить глазами за конкретным предметом;
- генерализованные эпилептические приступы различной продолжительности.
Важно понимать, что между спастическими мышечными сокращениями интервалы могут составлять менее 1 минуты. При этом частота припадков варьируется значительно, от нескольких десятков в день до сотни. При этом происходит сгибание и подергивание головы, руки вытягиваются, тело изгибается в дугу (заканчивается приступ полным расслаблением тела).
В процессе диагностики наши врачи уделают особое внимание симптомам Синдрома Веста, фиксируют каждую жалобу родителей, выясняют частоту, периодичность, характер приступов. Также проводится подробное изучение анамнеза, осмотр ребенка, оценивается специфика задержки психомоторного развития. После этого проводится ряд лабораторных и аппаратных исследований:
- общеклинические исследования крови и мочи;
- биохимическое исследование крови;
- электроэнцефалография;
- полисомнография; или магнитно-резонансная томография головного мозга.
Обязательным этапом диагностики являются консультации профильных специалистов, включая педиатра, детского невролога, эпилептолога.
Лечение Синдрома Веста
Тактика лечения в нашей клинике для каждого пациента разрабатывается индивидуально. Учитывая раннюю манифестацию и быстрое прогрессирование патологического процесса, особое значение уделяется ранней диагностике и лечению. Разрабатывая схему терапии, врачи «Вивантес» ориентируются на результаты диагностики, возраст пациента, специфику течения болезни, учитывают причины возникновения Синдрома Веста и множество других факторов.
Консервативное
В рамках консервативной терапии нашими специалистами применяются инновационные методики, используются препараты последнего поколения, длительность приема и дозировки которых рассчитаются индивидуально. К числу основных групп препаратов, направленных на лечение Синдрома эпилепсии Веста, относятся:
- миорелаксанты;
- глюкокортикостероиды;
- средства группы вальпроатов;
- адренокортикотропный гормон;
- витамины группы В6;
- иммуномодуляторы.
Хирургическое
Оперативные вмешательства в лечении Синдрома Веста имеют место в редких случаях, когда приступы обусловлены, например, опухолью головного мозга или туберозным склерозом. В таких случаях привлекаются хирурги высшей категории, которые проводят операции по удалению очагов поражения, опухолевых процессов. Максимальная точность и эффективность нейрохирургических методов обусловлена как квалификацией врачей, так и применением новейшего оборудования, например, роботизированной системы Da-Vinci.
Реабилитация
Реабилитационные программы, также разрабатывающиеся для пациентов индивидуально, рассчитаны на минимизацию или полное устранение приступов, а также всестороннее развитие ребенка. Для этого разрабатываются программы ЛФК, проводятся занятия с логопедами, дефектологами, психологами, назначается поддерживающая терапия.
Синдром веста клинические рекомендации
Синдром Веста или детские инфантильные спазмы относится к эпилептическим энцефалопатиям детского возраста, при которых эпилептиформная активность на электроэнцефалограмме (ЭЭГ) ведет к прогрессирующей психоневрологической дисфункции.
Как известно, Синдром Веста является зависимым от возраста эпилептическим синдромом с дебютом на первом году жизни, с 3-4 месяцев. Частота заболевания составляет 24–42 случая на 100 тыс. новорожденных. В 60% случаев страдают мальчики.
Синдром Веста имеет криптогенную и симптоматическую формы. Диагностическими критериями синдрома Веста являются особый тип эпилептических приступов – инфантильные спазмы, характерные изменения волн ЭЭГ в виде гипсаритмии и задержка психомоторного развития.
Причинами синдрома Веста могут самые разные заболевания и аномалии. Так среди заболеваний, приводящих к данному виду эпилепсии, обычно отмечают аномалии развития головного мозга, в том числе туберозный склероз, генетические мутации и внутричерепные кровоизлияния, характерные для недоношенных младенцев, и асфиксия. Однако чаще всего синдром Веста развивается у детей с диагнозом "асфиксия новорожденного", которая подразумевает кислородное голодание головного мозга младенца во время осложненных родов.
Синдром Веста чаще всего характеризуется тремя главными симптомами:
- Постоянные эпилептические приступы, которые почти не поддаются лечению.
- У детей наблюдается задержка психомоторного развития.
- На электроэнцефалограмме обнаруживаются типичные для данной болезни изменения – гипсаритмия.
Синдром Веста классифицируется врачами, как опасное психо-неврологическое заболевание, угрожающее не только здоровью, но и жизни ребенка. Каждый пятый ребенок с данным диагнозом умирает в возрасте до 5 лет. Наиболее выраженной причиной смерти являются врожденные аномалии развития головного мозга. Из оставшихся больных у 75% наблюдается нарушение психомоторного развития.
Менее 50% детей живут в стадии стойкой ремиссии, которая постоянно поддерживается приемом лекарственных средств. В 30% заболеваний болезнь поддается лечению. А 4% всех больных могут начать вести привычный образ жизни с восстановлением уровня физического и интеллектуального развития, который соответствует их возрасту.
Основная часть (до 90%) детей с подобным диагнозом обычно отстают в физическом и интеллектуальном развитии, даже после удачного избавления от судорог. Однако подобный прогноз связан не столько с самим заболеванием, сколько с первопричиной развития эпилепсии: аномалиями головного мозга, их размещения и степени проявления. Кроме того, постоянные сложные эпилептические приступы сами могут являться причиной разрушения мозга.
Крайне важно вовремя оценить состояние и поставить диагноз, для этого в ЦЕНТРЕ ДЕТСКОЙ НЕВРОЛОГИИ И ПСИХИАТРИИ Европейской клиники «Сиена-Мед» проводятся различные методы диагностических исследований:
- Электроэнцефалография (ЭЭГ)
- Видео - ЭЭГ (видео-электроэнефалография)
- Исследование мультимодальных вызванных потенциалов (ВП) головного мозга (зрительные ВП на реверсивный шахматный паттерн, акустические стволовые ВП, соматосенсорные ВП, Р300)
- Компьютерное исследование вегетативной нервной системы. Биохимические исследования, чтобы вовремя назначить соответствующее лечение
От сроков начала лечения зависит исход болезни. При адекватном подборе антиэпилептических препаратов можно достичь высоких результатов, в первую очередь, в прекращении приступов.
Поздняя диагностика и некорректные препараты могут лишь ухудшить течение синдрома Веста.
В ЦЕНТРЕ ДЕТСКОЙ НЕВРОЛОГИИ И ПСИХИАТРИИ Европейской клиники «Сиена-Мед» используются индивидуально подобранные методы лечения синдрома Веста у детей, воздействующие на все стороны патогенетического развития синдрома Веста у ребенка с учетом психопатологической структуры болезни, с обязательной корректировкой вегетативной нервной системы и нейромедиаторного обмена.
Синдром веста клинические рекомендации
9. Патогенетическая базовая гипотеза
- 1 фаза - цитопатическое повреждающее действием вируса на эндотелиальные клетки сосудов, которые несут на себе молекулы АПФ 2 и CD 147: вирус получает возможность взаимодействия с ними при разрушении аэро-гематического барьера и развивающейся виремии, воспалительная метаморфоза эндотелия приводит к активации системы свертывания крови по внутреннем механизму
- 2- фаза – при прогрессировании заболевания развивается гипреиммунная реакция («цитокиновый шторм»), оказывающий грубое и повсеместное повреждающее действие на эндотелий сосудов и обеспечивающий воспалительную реакцию с рекрутированием в очаг повреждения лейкоцитов, макрофагов, лимфоидных элементов и выраженной активацией свертывания крови («воспалительно-коагуляционного (тромботического) торнадо»). Генерализованная эндотелиопатия сопровождается выбросом высокомолекулярного фактора Виллебранда, стимулирующего активацию как плазменного, так и тромбоцитарного пути свертывания крови. Гиперэргическая иммунная реакция на SARS-CoV-2 у части больных, обусловливают бурное развитие иммунной воспалительной реакции, выраженного синдрома системной воспалительной реакции, ДВС, с тяжелой альтерацией ткани легких в виде диффузного альвеолярного повреждения, поражением других органов и тканей, с развитием картины септического шока.
- В июле 3 фаза - развитие диссеминированного системного васкулита с поражением сосудов мелкого и среднего калибра, тотального тромбоза, нарушение функции органов и систем (полиорганная недостаточность).
10 Вопросы классификации COVID-19 и постковидного синдрома
- Коронавирусная инфекция неуточненная (B34.2)
- Коронавирус как причина болезней, классифицированных в других рубриках (B97.2)
- Тяжелый острый респираторный синдром (ОРВИ) неуточненный (U04.9)
- Коронавирусная инфекция неуточненная (B34.2)
- COVID-19:
- лабораторно подтвержденный (позитивный, с указанием какими методами исследования – ПЦР, Ig)
- лабораторно не подтвержденный (негативный, с указанием какие методы подтверждения использовались и в какие сроки – ПЦР, Ig)
- с лабораторными признаками воспаления и внутрисосоудистой коагуляции крови (повышение скорости оседания эритроцитов, изменения в формуле крови, повышение уровня в крови СРБ, ферритина, Д-димера, фибриногена, интерлейкина 6, иных факторов растворимых комплекксов фибрин-мономеров, снижение активности антитромбина III, плазминогена, снижение уровня тромбоцитов)
- подтвержденный с использованием рентгенологического исследования (компьютерная томография)
- подтвержденный с помощью иных методов, включая данные искусственного интеллекта
- подтверждающие исследования не выполнялись
Для острой формы COVID-19 рекомендуется выделять следующие степени тяжести заболевания:
Легкая от практически отсутствующих симптомов болезни до кратковременных (от 1-2 до недели) изменений состояния в виде температурной реакции, кашля, слабости (возможно выраженная) и головной боли (возможно выраженная). Температура обычно в пределах 38ºС нет клинических признаков легочной недостаточности (одышки, нормальная сатурация крови кислородом – выше 93%), нет (если применимо) признаков поражения легочной ткани при рентгенологическом исследовании или при КТ, нет (если применимо) существенных изменений в лабораторных показателях воспаления (СРБ) и гемостаза
Средней тяжести – для определения средней степени тяжести болезни клиницисты должны учитывать разные факторы клинического, инструментального и лабораторного обследования как в сочетании, так и по отдельности: температура выше 38ºС в совокупности с выраженной слабостью и головной болью, чувство нехватки воздуха в сочетании с частотой дыхания более 22 в минуту и снижением сатурации ниже нормы (в пределах 95-93%), наличие повреждения легочной ткани при рентгенологическом исследовании или КТ до 25-30% поверхности (объема) легких, наличие клинических признаков поражения кишечника (диарея), высокие показатели острофазовых белков (СРБ, ферритин, фибриноген – если применимо), повышение уровня D-димера, выявляются признаки внутрисосоудистой коагуляции крови (повышение уровня растворимый комплексов фибрин-мономеров) при нормальном уровне тромбоцитов в крови, возможно снижение показателей фибринолитической активности крови. К среднетяжелым следует отнести пациентов с быстрой отрицательной динамикой заболевания.
Тяжелое течение инфекции считается при частоте дыхательных движений более 30 в мин., или SpO2 менее 93%,или PaO2 /FiO2 менее 300 мм рт.ст. Следует иметь ввиду отсутствие прямой корреляции одышки и напряжения показателей кислорода в крови. Обнаружение прогрессирования рентгенологических изменений в легких, типичных для вирусного поражения, по данным рентгенографии или КТ, в виде увеличение распространенности выявленных до 50% и более объема легочной ткани. К возможным признакам тяжелого течения следует отнести нарушения сознания, ажитацию, нестабильную гемодинамику (снижение АД менее 90/60 мм рт.ст. (или снижение на 45-50% от исходных величин)), наличие признаков преходящей парциальной почечной недостаточности (снижение диуреза менее 20 мл/час, повышение креатинина или мочевины) ДВС-синдром в тромботической фазе выражен: имеются лабильные изменения во всех звеньях системы свертывания крови – тромбоцитарном, плазменном, антикоагулянтном, фибринолитическом.
Крайне тяжелое состояние - все случаи развившейся органной недостаточности (необходимость респираторной поддержки, применения почечно-заместительной терапии), выраженный ДВС-синдром в геморрагической форме с феноменом потребления, сепсис, гемодинамический (септический) шок.
Реконвалесценции – при тяжелом течении болезни остаются многие проявления болезни, связанные с нарушением оксигенации (низкая сатурация, иногда – до 80%), слабость, субфебрильная температура; постепенно нормализуются лабораторные показатели воспаления (если применимо), рентгенологическая картина повреждения легких (если применимо); период реконвалесценции может занимать длительное время и его не стоит путать с длительным течением болезни или постковидным синдромом.
Классификации постковидного синдрома по клиническим проявлениям:
• Постковидный тромбоваскулит различных отделов нервной системы (постковидный менингоэнцефалит)
- с поражением центральной, периферической и вегетативной нервной системы (в том числе двигательные и чувствительные нарушения) – выраженная слабость, нарушения зрения и слуха, когнитивные нарушения (расстройства памяти, внимания, трудности концентрации), эмоционально-поведенческие расстройства (тревога, депрессия, делирий, нарушения сна,), нарушения мозгового кровообращения (артериальные и венозные инфаркты, церебральные кровоизлияния с клиникой инсульта или без), другими множественными проявлениями (см. Раздел Образ болезни)
- с поражением кардиоваскулярного сегмента метасимпатической нервной системы (сердечно-сосудистые проявления, нарушение функции периферического сердца (М.В.Яновский)) – нарушение регуляции АД, необъяснимая, в том числе постуральная тахикардия
- с поражением энтерального сегмента метасимпатической нервной системы (регуляции пищеварительной системы) – диарея, реже – боли в животе спастического характера
- с дыхательной дисфункцией – нарушения акта дыхания (саккадированное дыхание), чувство заложенности грудной клетки
- с дисфункцией мочеотделения – учащенное мочеиспускание, позывы к мочеиспусканию
- с гормональной дисфункцией – изменения гормонального статуса щитовидной железы, нарушения сахарного обмена, иногда, при наличии диабета в анамнезе – развитие неконтролируемой гипергликемии, нарушения менструального цикла, нарушения половых функций у мужчин (либидо, эрекции)
- с дисфункцией высшей нервной деятельности (в том числе – депрессии, когнитивные расстройства, панические атаки, суицидальные мысли)
• Ипохондрический вариант постковидного синдрома – выраженное беспокойство относительно своего состояния с постоянным ожиданием смерти, развития необратимых осложнений (инсульт, инфаркт), чрезвычайная озабоченность своим здоровьем, навязчивыми и катастрофическими жалобами (часто нетипичные и разнообразные, причудливые – ощущения движения крови или пузырьков по сосудам, распирание головы с ощущением, что на вот-вот лопнет, внутренняя дрожь, как трансформатор в теле и тд.), предположение, что, кроме основного заболевания, проявляющегося видимыми симптомами, есть какое-то дополнительное; при этом уверен, что знает лучше, какое у него «на самом деле» заболевание (важный признак) и сомневается и не приемлет рекомендацияй (иногда негативен в своих оценках рекомендаций, особенно – если обсуждается психиатрическая компонента заболевания),обычно имеется чрезвычайно большое число разнообразных дополнительных исследований и консультаций, социальная дезадоптация, образование порочного круга симптомов и агрессивных методов лечения, когда осложнения последних усугубляют имеющуюся симптоматику.
• Постковидный васкулит микро- и макрососудов в бассейне кожи и ее придатков – различные полиморфные высыпания на коже, включая синячковость и васкулитоподобные изменения, мраморность кожи, разнообразные формы синдрома Рейно, аллергические дерматиты (преходящая крапивница), выпадение волос, линии Бо (поперечная исчерченость ногтей); сюда можно отнести сосудистую сеточку (ретикулярная асфиксия), сетчатое ливедо, телеангиоэктазии и аневризматические изменения вен с появлением видимых глазом «узелков» на периферических венах.
• Вторичные функционально-морфологические изменения тканей и систем (легочная, почечная, печеночная недостаточность, последствия тромбоза глубоких вен нижних конечностей, инсульта, инфаркта миокарда, тромбоэмболии легких)
• Отдельные синдромы аутоиммунных реакций: синдром Гийена-Барре, синдром Миллера-Фишера,Кавасаки-подобный синдром и др.
Постковидный синдром может быть
• с лабораторным или инструментальным подтверждением эпизода острого COVID-19
• с лабораторным подтверждением воспаления, внутрисосудистой коагуляции крови, наличие иных маркеров, например, изменений гормонального статуса
• с инструментальным подтверждением (морфологические и функциональные маркеры изменения мозговых тканей и функций)
• без лабораторного и инструментального подтверждения
Классификация эпилепсии
Эпилепсия — заболевание головного мозга, соответствующее любому из следующих состояний:
- Не менее двух неспровоцированных (или рефлекторных) эпилептических приступов с интервалом > 24 ч.
- Один неспровоцированный (или рефлекторный) эпилептический приступ и вероятность повторных приступов, соответствующая общему риску рецидива (³ 60 %) после двух неспровоцированных эпилептических приступов, в следующие 10 лет.
- Диагноз эпилептического синдрома.
Критерии разрешения эпилепсии включают достижение определенного возраста у пациентов с зависящим от возраста эпилептическим синдромом либо отсутствие эпилептических приступов в течение 10 лет у пациентов, не получавших противосудорожные препараты более 5 лет.
Классификация эпилепсии (Международная противоэпилептическая лига (ILAE) 2017 г. )
Классификация эпилепсии проводится после определения критериев диагностики эпилепсии (определение выше). Классификация проводится с использованием трехуровнего ранжирования — определение типа приступов, типа эпилепсии и синдрома эпилепсии. Нейроимиджинг, ЭЭГ и другие исследования, если они есть, помогают улучшить классификацию на всех трех уровнях. Где это возможно, следует установить диагноз на всех трех уровнях. Этиологию эпилепсии следует устанавливать с самого начала и на каждом этапе всего диагностического пути. Знание этиологии может способствовать оптимизации классификации и имеет важные лечебные последствия для пациента.
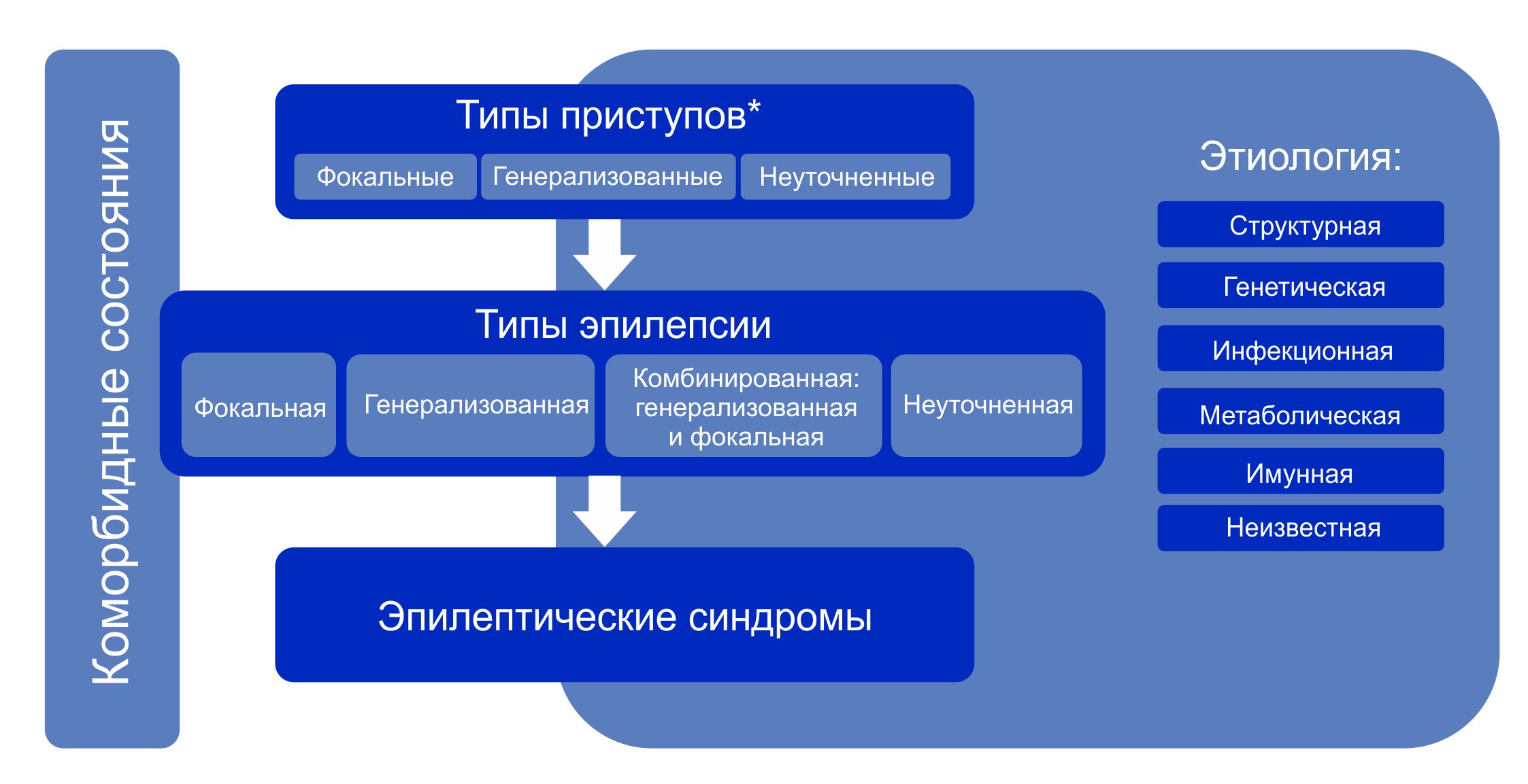
Структура Классификации эпилепсии ILAE 2017 г.
Примечание. * Оценивается по началу приступа.
Авакян Г. Н. Блинов Д. В. Лебедева А. В. Бурд С. Г. Авакян Г. Г. Классификация эпилепсии Международной Противоэпилептической Лиги: пересмотр и обновление 2017 года. Эпилепсия и пароксизмальные состояния. 2017; 9 (1): 6–25. DOI: 10.17749/2077–8333.2017.9. 1.006–025.
Алгоритм классификации эпилепсии:
- На первом этапе (уровне) определяют тип приступа: фокальный, генерализованный или с неизвестным началом.
- На втором этапе (уровне) устанавливают тип эпилепсии: фокальная, генерализованная или сочетанная фокальная и генерализованная, или неизвестная (unknown).
- На третьем этапе (уровне) определяют эпилептический синдром и коморбидность. Эпилептический синдром представляет собой совокупность характеристик, включая тип приступа, данные ЭЭГ и нейровизуализации, часто имеет возрастзависимый характер, провоцирующие факторы, хронозависимость и в ряде случаев определенный прогноз.
- На четвертом этапе (уровне) устанавливают этиологию эпилепсии: структурная, генетическая, инфекционная, метаболическая, иммунная или с неизвестной этиологией.
Генерализованная эпилепсия
Пациенты с генерализованной эпилепсией имеют генерализованные типы приступов и могут иметь типичные интериктальные и / или иктальные ЭЭГ паттерны, которые сопровождают генерализованные типы приступов (например, генерализованный спайк-волна). Сопутствуют семейная история генерализованных типов приступов или генерализованная эпилепсия.
Генетическая / идиопатическая генерализованная эпилепсия представляет собой эпилепсию с генерализованными приступами, ассоциированнами с генерализованными эпилептиформными паттернами ЭЭГ, такими как генерализованная спайк-волновая активность, которая, считается, имеет генетическую этиологию. Однако это не всегда означает, что эти эпилепсии унаследованы или могут передаваться потомству, поскольку генетическая этиология может быть спонтанной новой мутацией или наследование может быть сложным. Таким образом, термин «идиопатическая генерализованная эпилепсия» используется как синоним генетической генерализованной эпилепсии, и клиницист может выбрать, какой термин использовать, в зависимости от важности акцента на генетическом наследовании для конкретного пациента. Генетические / идиопатические генерализованные эпилепсии включают детскую абсансную эпилепсию, юношескую абсансную эпилепсию, юношескую миоклоническую эпилепсию, и эпилепсию с генерализованными тонико-клоническими приступами.
Фокальная эпилепсия
Пациенты с фокальной эпилепсией имеют фокальные типы приступов и могут иметь типичные интериктальные и / или иктальные ЭЭГ-паттерны, которые сопровождают фокальные типы приступов (такие как фокальные острые волны или фокальное интериктальное замедление). Нейроимиджинг демонстрирует фокальную структурную аномалию мозга и способствует установке диагноза, хотя пациенты с генетической этиологией и нормальной визуализацией могут также иметь фокальную эпилепсию. Фокальные эпилепсии могут быть унифокальными, мультифокальными или полушарными.
Сочетанная генерализованная и фокальная эпилепсия
Пациенты могут иметь как генерализованные, так и фокальные типы приступов, с интериктальными и / или иктальными ЭЭГ-паттернами, которые сопровождают оба типа приступов. Пациенты с синдромом Драве и синдромом Леннокс-Гасто могут иметь генерализованную и фокальную эпилепсию.
Неизвестная эпилепсия
Термин «неизвестный» используется для обозначения в случае, когда понимается, что у пациента есть эпилепсия, но невозможно определить, является ли она фокальной, генерализованной или комбинированной: фокальной и генерализованной. Это бывает при недостаточной информации для классификации эпилепсии, например, если ЭЭГ является нормальной / неинформативной.
Синдромы эпилепсии
В то время как концептуализация эпилепсий по их этиологии очень важна, эпилепсии также могут быть организованы (в соответствии с установленными достоверными общепринятыми клиническими и ЭЭГ — характеристиками) в эпилептические синдромы. Такие синдромы имеют типичный возраст начала приступа, специфические типы приступов и характеристики ЭЭГ и часто другие признаки, которые вместе взятые позволяют диагностировать конкретный эпилептический синдром. Идентификация синдрома эпилепсии полезна, так как она предоставляет информацию о том, какие основные этиологии следует учитывать и какие антиконвульсанты могут быть наиболее полезными. Некоторые эпилептические синдромы демонстрируют аггравацию приступов при определенных антиконвульсантах, чего можно избежать при ранней диагностике синдрома эпилепсии.
Эпилептические синдромы
Неонатальный и младенческий период:
- самокупирующиеся неонатальные приступы и самокупирующаяся семейная неонатальная эпилепсия;
- самокупирующаяся семейная и несемейная младенческая эпилепсия;
- ранняя миоклоническая энцефалопатия;
- синдром Отахара;
- синдром Веста;
- синдром Драве;
- миоклоническая эпилепсия младенчества, младенческая эпилепсия с мигрирующими фокальными приступами;
- миоклоническая энцефалопатия при непрогрессирующих заболеваниях;
- фебрильные приступы, фебрильные приступы плюс, генетическая эпилепсия с фебрильными приступами плюс.
Детский возраст:
- эпилепсия с миоклонически-атоническими (ранее – астатическими) приступами;
- синдром Леннокса–Гасто;
- фебрильные приступы, фебрильные приступы плюс;
- абсансная эпилепсия;
- эпилепсия с миоклоническими абсансами;
- детская затылочная эпилепсия с ранним дебютом (синдром Панайотопулоса);
- детская затылочная эпилепсия с поздним дебютом (синдром Гасто);
- фотосенситивная затылочная эпилепсия;
- самокупирующаяся эпилепсия с центрально-темпоральными спайками;
- атипичная эпилепсия с центрально-темпоральными спайками;
- эпилептическая энцефалопатия с продолженной пик-волновой активностью во время сна;
- синдром Ландау–Клеффнера;
- аутосомно-доминантная ночная лобная эпилепсия.
Подростковый и взрослый возраст:
- юношеская абсансная эпилепсия;
- юношеская миоклоническая эпилепсия;
- эпилепсия с изолированными генерализованными тонико-клоническими приступами;
- аутосомно-доминантная эпилепсия со слуховыми проявлениями;
- другие семейные височные эпилепсии.
Любой возраст:
- семейная фокальная эпилепсия с вариабельным фокусом;
- рефлекторные эпилепсии;
- прогрессирующие миоклонические эпилепсии.
Этиология эпилепсии
В последние годы значительно расширилось наше понимание основополагающих причин эпилепсии, подкрепленное достижениями в области современной нейровизуализации и генетического тестирования. Такая терминология, как «идиопатическая», «криптогенная» и «симптоматическая», больше не используется. Эпилепсии теперь описываются более точно конкретными основополагающими причинами.
Генетическая этиология
Понятие генетической эпилепсии заключается в том, что эпилепсия, насколько мы понимаем, является прямым результатом известного или предполагаемого генетического дефекта (ов), в котором судороги являются основным симптомом расстройства. Генетический дефект может возникать на хромосомном или молекулярном уровне. Важно подчеркнуть, что «генетический» не означает то же, что «унаследовано», поскольку мутации de novo не являются редкостью. Наличие генетической этиологии не исключает экзогенного влияния на возникновение эпилепсии.
Наиболее важные генетические причины эпилепсии, которые могут быть идентифицированы при клиническом тестировании:
- хромосомные аномалии;
- аномалии гена.
Существует много способов, которыми генетические факторы могут способствовать развитию эпилепсии. Определенные генетические факторы, возможно, не были унаследованы и не могут быть переданы потомству. Вот некоторые важные генетические концепции, используемые на этом веб-сайте, и их определения:
- унаследованные аномалии генов, аутосомно-доминантное, аутосомно-рецессивное и менделевское наследование;
- приобретенные аномалии генов — de novo, спорадические, мозаицистические, зародышевые и соматические;
- полигенная / комплексная генетическая этиология.
Структурная этиология
Структурные эпилепсии определяются как имеющие выраженную структурную аномалию мозга, которая связана с существенно повышенным риском эпилепсии. Структурная аномалия мозга может быть приобретена (например, вследствие инсульта, травмы или инфекции) или может быть генетического происхождения; однако, как мы это понимаем в настоящее время, структурная аномалия мозга представляет собой отдельное нарушение, расположенное между приобретенным или генетическим дефектом и эпилепсией.
Нейроимиджинг
Магнитно-резонансная томография (МРТ) с использованием 1.5 Тесла аппарата является минимальным стандартным исследованием для исключения структурной аномалии. При этом исследовании важно выполнять протоколы, специфические для эпилепсии, которые позволяют тщательно изучать специфические приобретенные аномалии (например, склероз гиппокампа ) и тонкие пороки развития коры головного мозга, такие как фокальная дисплазия коры. Изображение с использованием 3 Тесла и использование передовых методов анализа программного обеспечения может помочь в выявлении структурных нарушений, не очевидных при обычной МРТ. Интериктальная и иктальная ЭЭГ вместе с дополнительными функциональными исследованиями нейровизуализации, такими как ПЭТ, ОФЭКТ и МЭГ, помогают внимательно изучить конкретную область мозга и идентифицировать тонкую аномалию. У детей младшего возраста в возрасте до 2 лет тонкие аномалии не могут быть выявлены из-за незаконченной миелинизации, и повторное исследование требуется в динамике.
Общие структурные аномалии мозга, связанные с эпилепсией:
- пороки развития коры головного мозга;
- сосудистые пороки развития;
- гиппокампальный склероз;
- гипоксически-ишемические структурные аномалии;
- травматическая повреждение мозга;
- опухоли;
- порэнцефалическая киста.
Метаболическая этиология
Метаболические эпилепсии определяются как имеющие определенное метаболическое нарушение, связанное с выраженным риском развития эпилепсии. Метаболические расстройства имеют генетическое происхождение; однако, как мы это понимаем в настоящее время, метаболические аномалии представляют собой отдельное нарушение, стоящее между генетическим дефектом и эпилепсией.
Важные метаболические эпилепсии:
- дефицит биотинидазы и голокарбоксилазы-синтазы;
- дефицит церебрального фолата;
- нарушения креатина;
- приступы при нарушении фолатного цикла;
- недостаточность транспортера глюкозы 1 (GLUT1);
- митохондриальные расстройства;
- пероксисомальные расстройства;
- пиридоксинзависимая эпилепсия.
Иммунная этиология
Иммунные эпилепсии определены как имеющие выраженную иммунную опосредованную этиологию с подтверждением воспаления центральной нервной системы, что, как было показано, связано с существенно повышенным риском развития эпилепсии.
Важные иммуноопосредованные эпилепсии:
- Синдром Расмуссена;
- Антителоопосредованная эпилепсия.
Инфекционная этиология
Наиболее распространенная этиология эпилепсии во всем мире является инфекционной, особенно в развивающихся странах. Инфекции в центральной нервной системе могут вызывать как острые симптоматические припадки (которые тесно связаны со сроками первичной инфекции), так и эпилепсией. Инфекционная этиология включает туберкулез, ВИЧ, церебральную малярию, нейроцистицеркоз, подострый склерозирующий панэнцефалит, церебральный токсоплазмоз. Эти инфекции иногда имеют структурный коррелят, однако основная причина эпилепсии определяется как инфекционный процесс. Инфекционная этиология может иметь специфические последствия лечения. Существуют также последствия для общественного здравоохранения, поскольку профилактика таких инфекций может снизить нагрузку на эпилепсию, особенно в развивающихся странах. Наиболее распространенные из таких инфекций следующие:
- бактериальный менингит или менингоэнцефалит;
- церебральная малярия;
- церебральный токсоплазмоз;
- цитомегаловирусная инфекция;
- ВИЧ;
- нейроцистицеркоз;
- туберкулез;
- вирусный энцефалит;
- подострый склерозирующий панэнцефалит;
- прочие инфекции (токсокариоз, шистосомоз, болезнь Лайма (нейроборрелиоз).
Неизвестная этиология
«Неизвестная» этиология должна рассматриваться нейтрально и обозначать, что природа основной причины возникновения эпилепсии пока неизвестна; это может быть фундаментальный генетический дефект или отдельное, пока еще установленное, нарушение.
Имитаторы эпилепсии
Существует ряд состояний, связанных с рецидивирующими пароксизмальными событиями, которые могут имитировать симптомы, и ошибочно диагностироваться как эпилепсия. Важно, чтобы эти расстройства учитывались при оценке пароксизмальных событий, так как частота ошибочных диагнозов при эпилепсии высока во всем мире. История заболевания и видеозапись приступов помогают установить правильный диагноз. Существуют некоторые состояния, при которых могут сосуществовать эпилептические и неэпилептические события.
Предикторы неблагоприятного исхода синдрома Веста

Актуальность. Синдром Веста — младенческая эпилептическая энцефалопатия, характеризующаяся триадой симптомов: инфантильные спазмы, изменения на электроэнцефалограмме (ЭЭГ) в виде гипсаритмии и задержка психомоторного развития. Определение предикторов исхода заболевания даст возможность быстро подобрать оптимальную терапию, определить сроки динамического наблюдения и улучшить результаты лечения.
Цель работы — выявить предикторы исхода синдрома Веста.
Материалы и методы. В исследование вошли 132 пациента, которые проходили лечение с 2000 по 2018 г. Возраст детей на момент начала наблюдения составлял 5 мес. – 17 лет 11 мес. Возраст дебюта спазмов варьировал от 1-х суток жизни до 3 лет 2 мес. По этиологическому критерию пациентов разделили на три группы: 1-я группа — пациенты со структурной формой (60 пациентов; 45,5 %), 2-я группа — пациенты с генетической формой (39 детей; 29,5 %), 3-я группа — пациенты с невыясненной этиологией заболевания (33 ребенка; 25,0 %). При оценке терапии внимание уделяли эффективности препаратов первых трех линий, проводимой гормональной терапии, а также дальнейшему подбору антиэпилептических препаратов.
Результаты. Эпилептические спазмы купированы у 76 (57,6 %), все приступы — у 48 (36,4 %) детей. У пациентов 3-й группы отмечена бóльшая частота купирования спазмов (87,9 % против 48,7 и 46,7 % соответственно) и полной ремиссии (72,7 против 26,7 и 27,6 % соответственно), чем в 1-й и 2-й группах. Позитивное прогностическое значение для купирования спазмов имели: нормальное нервно-психическое развитие до дебюта спазмов, отсутствие эпиактивности или наличие региональной эпиактивности на ЭЭГ в динамике, наличие диффузных изменений на МРТ. Негативное прогностическое значение имели: неонатальные судороги, наличие эпиактивности на ЭЭГ и очагового дефицита до спазмов, наличие других приступов, кроме спазмов, патология зрения и слуха, необходимость применения ≥2 препаратов. У пациентов, достигших ремиссии, отмечена лучшая компенсация моторного и психоречевого развития.
Выводы. Предикторами неблагоприятного исхода синдрома Веста можно считать: структурную и генетическую форму заболевания, неонатальные судороги, наличие эпилептиформной активности (ЭА) на ЭЭГ, нарушение нервно-психического развития и наличие очаговой патологии до спазмов, наличие других приступов кроме спазмов, сохранение ЭА в динамике, неэффективность терапии первой линии.
Ключевые слова
Полный текст
Актуальность
Синдром Веста является одной из катастрофических эпилепсий детского возраста вследствие сложности контроля приступов и задержки умственного развития [14]. Впервые данное заболевание было описано английским педиатром W.J. West в 1841 г. в журнале Lancet на примере болезни собственного сына [2, 3]. Синдром Веста — младенческая эпилептическая энцефалопатия, характеризующаяся триадой симптомов: инфантильные спазмы, изменения на электроэнцефалограмме (ЭЭГ) в виде гипсаритмии и задержка психомоторного развития. Однако диагноз может быть установлен и при наличии двух признаков из трех [2, 3, 10]. Инфантильные спазмы — быстрые сокращения мышц продолжительностью 1–2 с, занимающие по своей скорости промежуточное положение между миоклоническими и тоническими приступами [1, 3]. Эпилептический спазм — более широкий термин, не тождественный синдрому Веста. Спазмы могут быть вне синдрома Веста в любом возрасте [1]. Согласно литературным данным, ряд зарубежных исследователей (Riikonen R., Yilmaz S., Nikolic D., Hamano S., Karvelas G., Mohamed B.P. et al.) старались выявить прогностически значимые факторы. Однако при анализе этих работ нами отмечены малая выборка пациентов, их неоднородность и преимущественно небольшой срок наблюдения. Кроме того, в данных работах имеются значительные различия в перечне наиболее важных прогностических признаков [4, 5, 7–9, 11–13, 15]. Таким образом, определение предикторов даст возможность прогнозировать исход синдрома Веста уже при первом клиническом осмотре пациента, определить выбор оптимальной терапии, сроки динамического наблюдения и улучшить результаты лечения.
Цель работы — определить прогностически значимые факторы исхода синдрома Веста, в том числе оценить зависимость исхода от этиологии заболевания, исследовать влияние эпилептических спазмов и других видов приступов на психомоторное развитие ребенка и развитие аутистикоподобного синдрома.
Материалы и методы
В исследование вошли 132 пациента, которые проходили стационарное лечение и наблюдались амбулаторно в условиях Нижегородской областной детской клинической больницы, Детской городской клинической больницы № 1 Нижнего Новгорода и Института детской и взрослой неврологии и эпилепсии имени святителя Луки Москвы с 2000 по 2018 г. Ранний анамнез у ряда пациентов оценивали по данным медицинской документации при анализе историй болезни и амбулаторных медицинских карт. У всех пациентов когда-либо регистрировали эпилептические спазмы. Среди вошедших в исследование детей 74 — мужского пола, 58 — женского. Таким образом, распределение детей по полу составило 1,28 : 1. Возраст детей на момент начала наблюдения составлял 5 мес. — 17 лет 11 мес. (средний 5 лет 9 мес. ± 4 года 5 мес.).
При оценке данных анамнеза всех пациентов разделили на три группы: 1-я группа — пациенты со структурной формой заболевания (60 пациентов; 45,5 %), 2-я группа — пациенты с генетической формой заболевания (39 детей; 29,5 %), 3-я группа — пациенты с невыясненной этиологией заболевания (33 ребенка; 25,0 %).
Возраст дебюта спазмов варьировал от 1-х суток жизни до 3 лет 2 мес. Ранний дебют эпилептических спазмов (до 3 мес.) наблюдали у 17 (12,9 %) детей, что не исключает наличие у ребенка других форм эпилепсии с последующей трансформацией в синдром Веста. Классический дебют (в возрасте 3–12 мес.) отмечали у 101 (76,5 %) ребенка, поздний дебют (в возрасте ≥12 мес.) — у 14 (10,6 %) детей. Пик дебюта спазмов приходится на возраст 4–6 мес.
У 125 (94,7 %) детей регистрировали серийные спазмы, у 7 (5,3 %) — одиночные. В 76 (57,6 %) случаях отмечали симметричные спазмы, в 56 (42,4 %) случаях — асимметричные. У 81,7 % детей выявляли флексорные, у 11,7 % — экстензорные и у 6,6 % — смешанные спазмы. В табл. 1 отражены дополнительные данные анамнеза пациентов сравниваемых групп.
Данные анамнеза пациентов
Читайте также: